In the search to find increasingly more effective therapeutics, core hopping, or scaffold hopping, has been a key approach among drug discovery teams for several years. Offering an economical route to both hit finding and lead optimization, scaffold hopping enables faster exploration of new IP space and the modulation of critical properties when smaller modifications of peripheral moieties are unable to achieve the desired outcomes.
Varied computational approaches have been developed to increase the efficiency of scaffold hopping pipelines, but reliable assessment of core libraries has remained largely elusive. This creates a burden on drug discovery teams due to the significant investment of time and resources required for the synthesis of small molecules with new cores.
In TandemViz, you can generate a core hopping library with just a few simple clicks and smoothly transition to accurately assessing binding affinity and other pharmacokinetic properties. By fast-tracking your drug discovery timeline, you’re empowered to explore more of the chemical space than ever before.
While a powerful tool in computational drug discovery, traditional Free Energy Perturbation (FEP) approaches for calculating binding affinity, have proved much less effective for predictions across small molecules with different cores. With our revolutionary CBFE technology, we’ve made it possible to digitally assay structurally dissimilar compounds from core hopping with the accuracy of RBFE FEP calculations and at significantly less computational cost than ABFE.
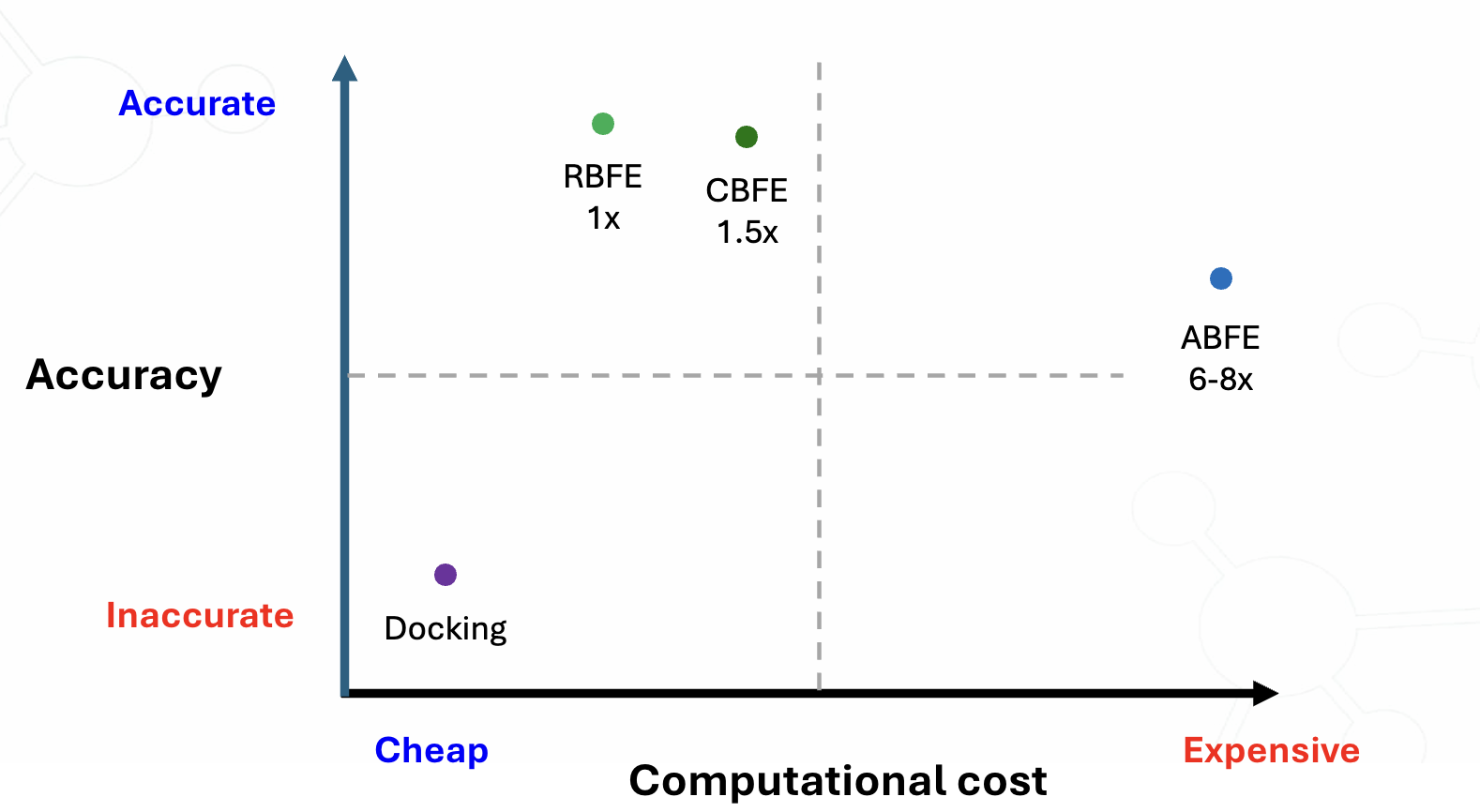
Integrated with our full platform through TandemViz, you can leverage our entire core hopping workflow in one collaborative workspace. Harness the power of generative AI to create a library of 100s of cores in TandemGen and seamlessly calculate the binding free energies of these molecules within 10- to 20-fold of a biological assay with CBFE in TandemFEP.
To begin your core hopping enumeration in TandemGen, you first select a scaffold by sketching a molecule with a core to replace or by searching for a molecule within your project as a starting point. Our interface allows you to visualize your reference molecule in the way that works for you, as either a 2D or 3D model with multiple drawing options.
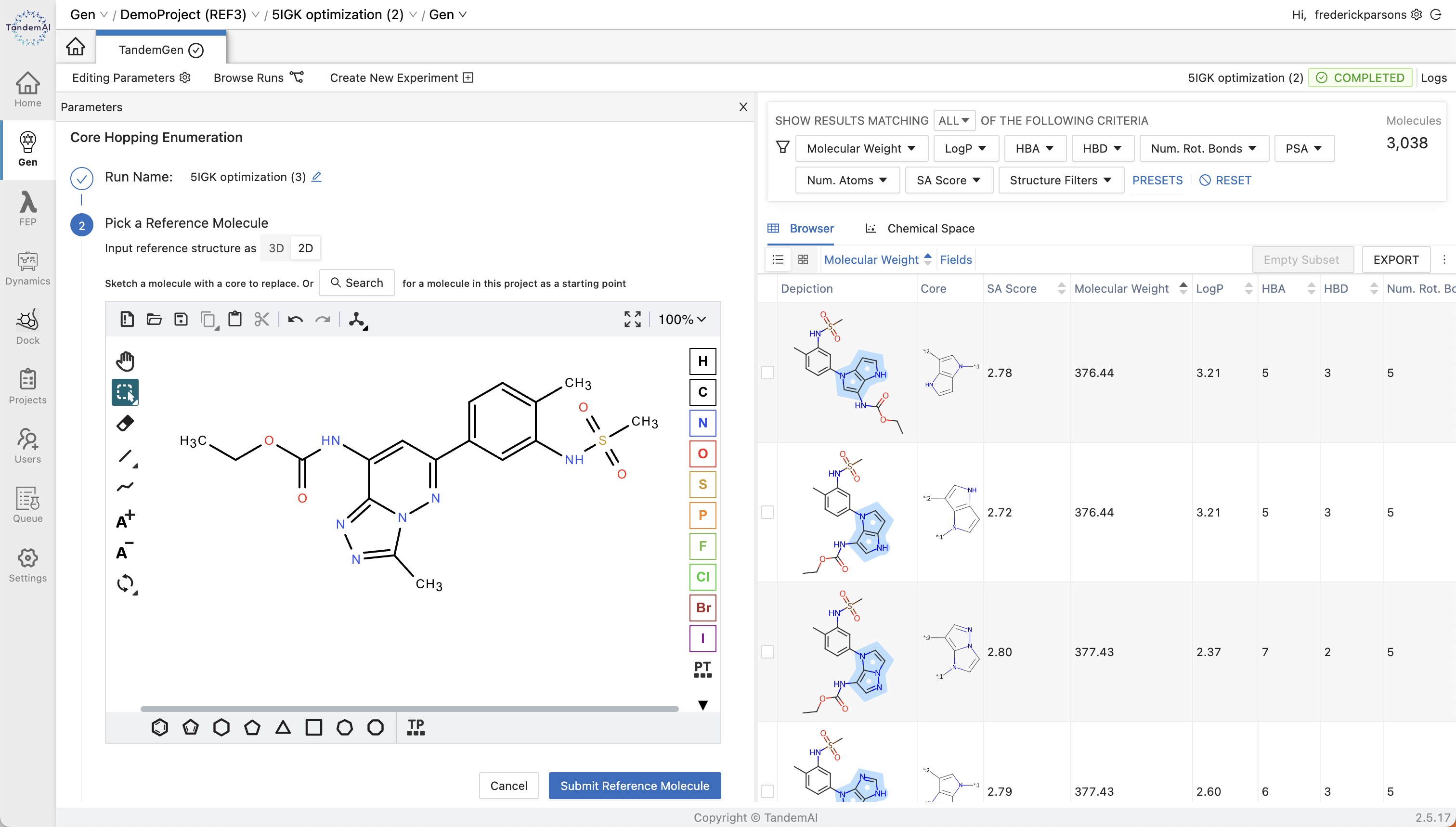
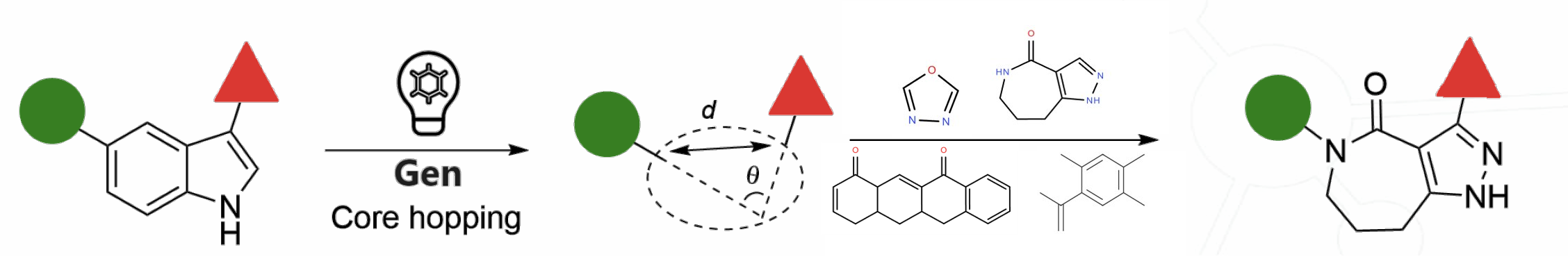
You can then identify the core within your scaffold that you want to replace by selecting the two bonds on either side. The ‘Generate Structures’ button can then be used to create a new library for core hopping that can be triaged or exported to other applications from within TandemGen.
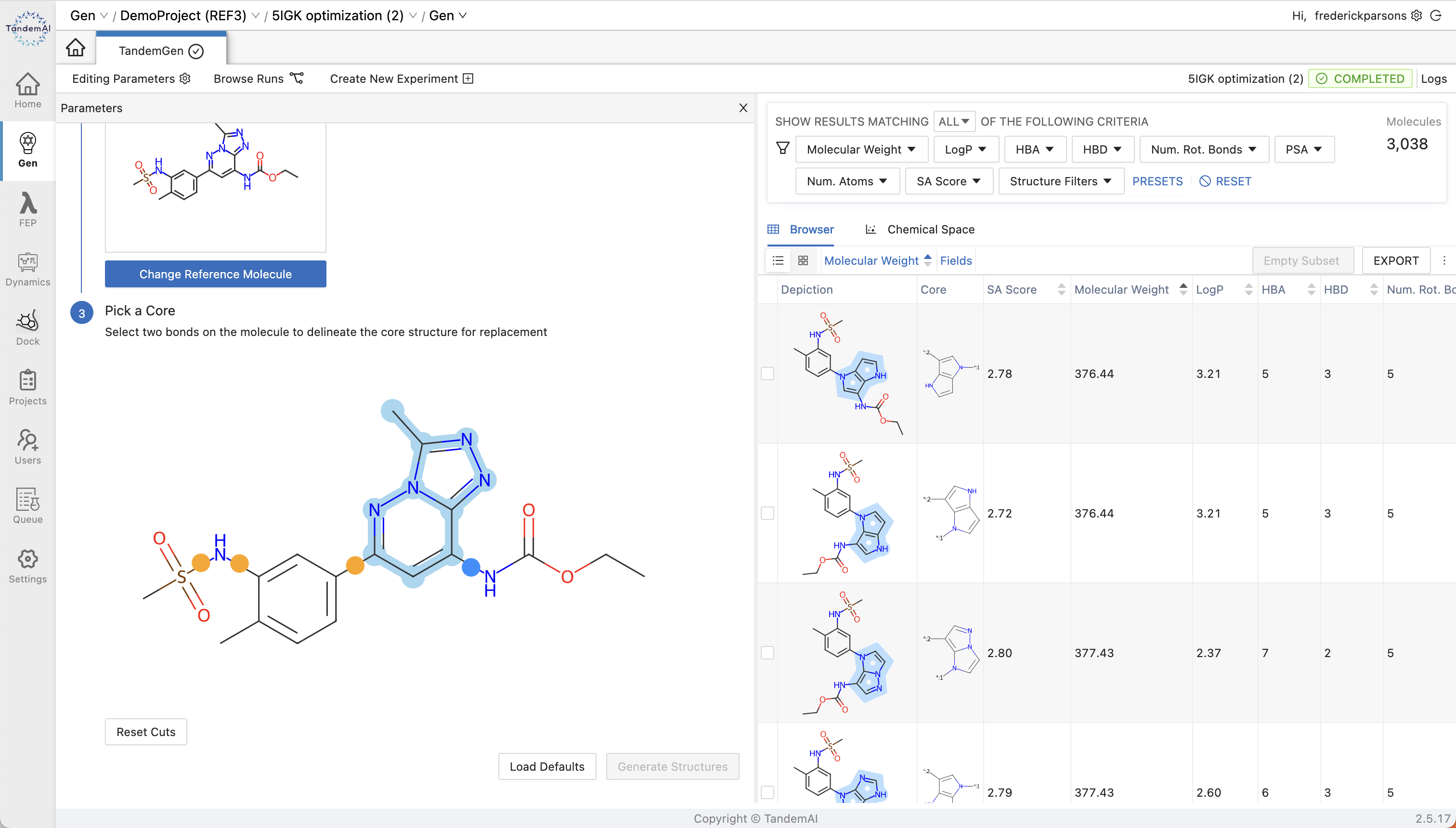
With TandemGen, you don’t need to worry about replacement cores being structurally and chemically reasonable. Our geometry-based approach evaluates the distance and angle between substituents to ensure that alternative cores maintain similar R-group orientations for retained functionality.
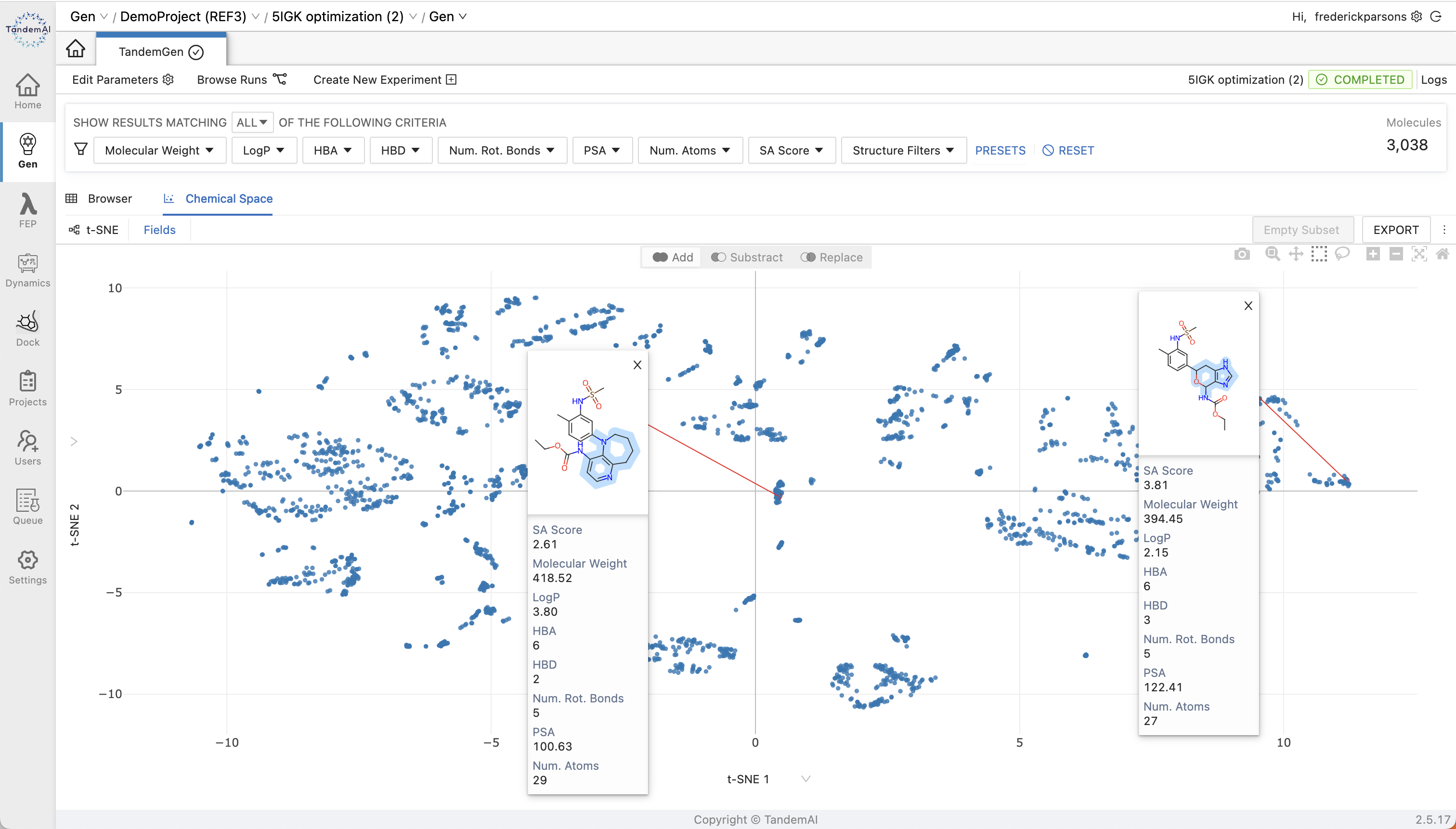
Once your library has been generated, the user-friendly TandemViz interface makes exploring your enumerated cores effortless. You can view your results either across the chemical space or in the browser, and use real-time filtering, sorting, and plotting features, including t-SNE plots, to easily identify promising candidates for further evaluation.
 Learn more in our interactive release showcase
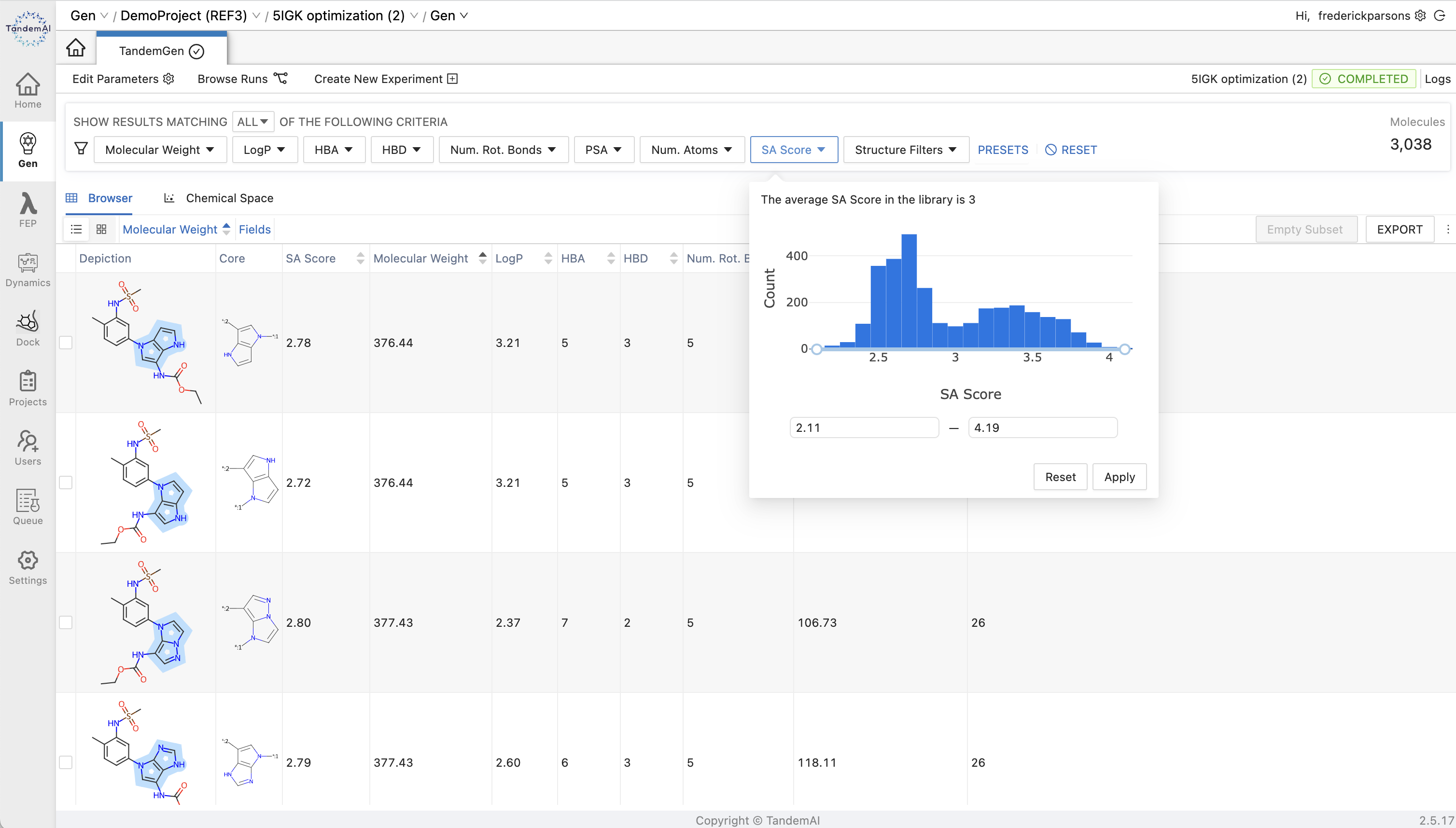
Learn more in our interactive release showcaseNarrow your results to cores with the properties that matter the most to you with the extensive array of filters available in TandemGen. Synthetic accessibility scores for your entire library are available as an interactive plot for you to filter by and future-proof your workflow against potential challenges at the synthesis stage.
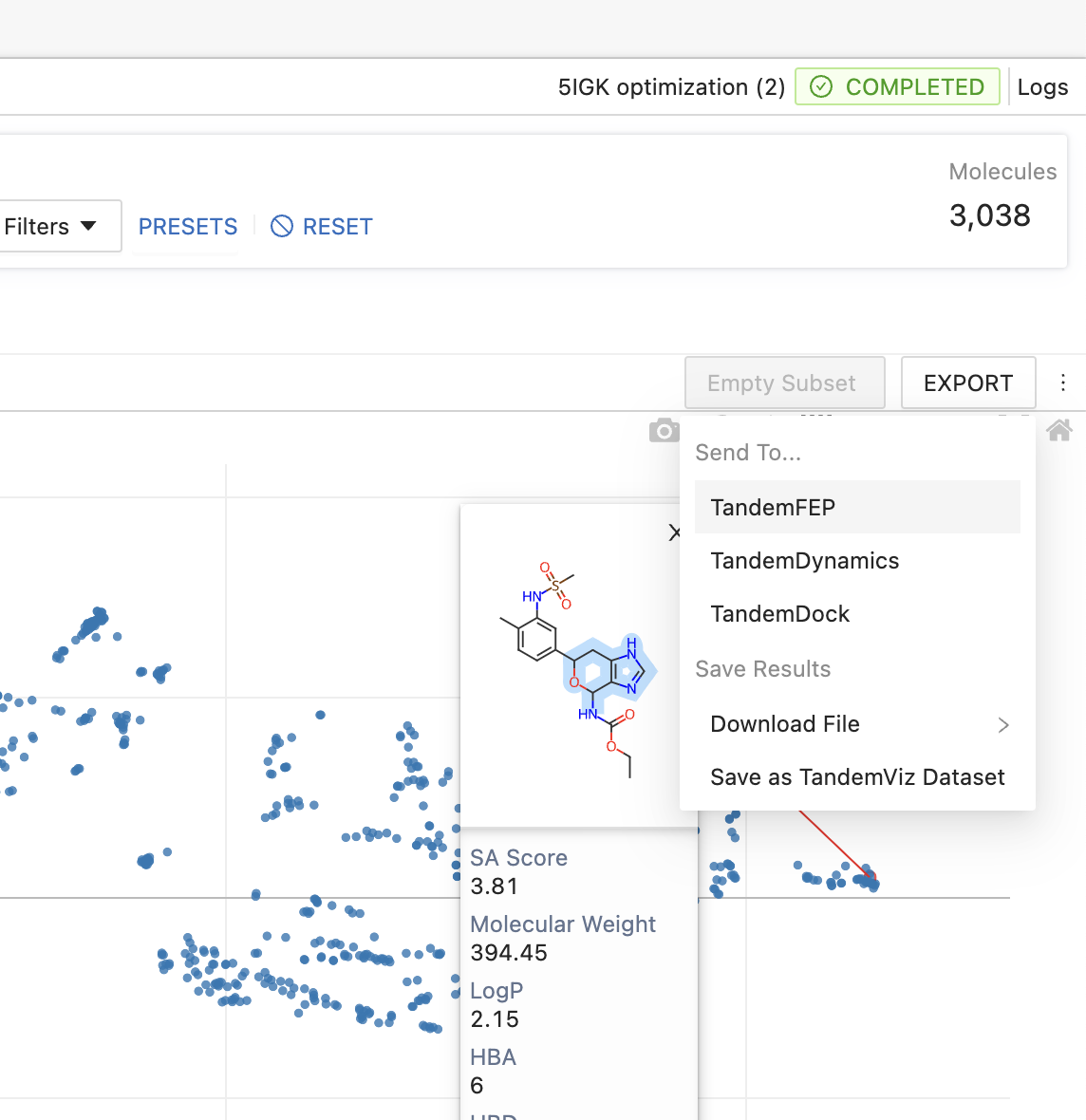
Since TandemViz is a highly connected platform, selecting a subset of candidates and sending them for further evaluation is a snap. Whether you want to move to CBFE in Tandem FEP or explore other properties in TandemDock and TandemDynamics, exporting is quick and seamless.
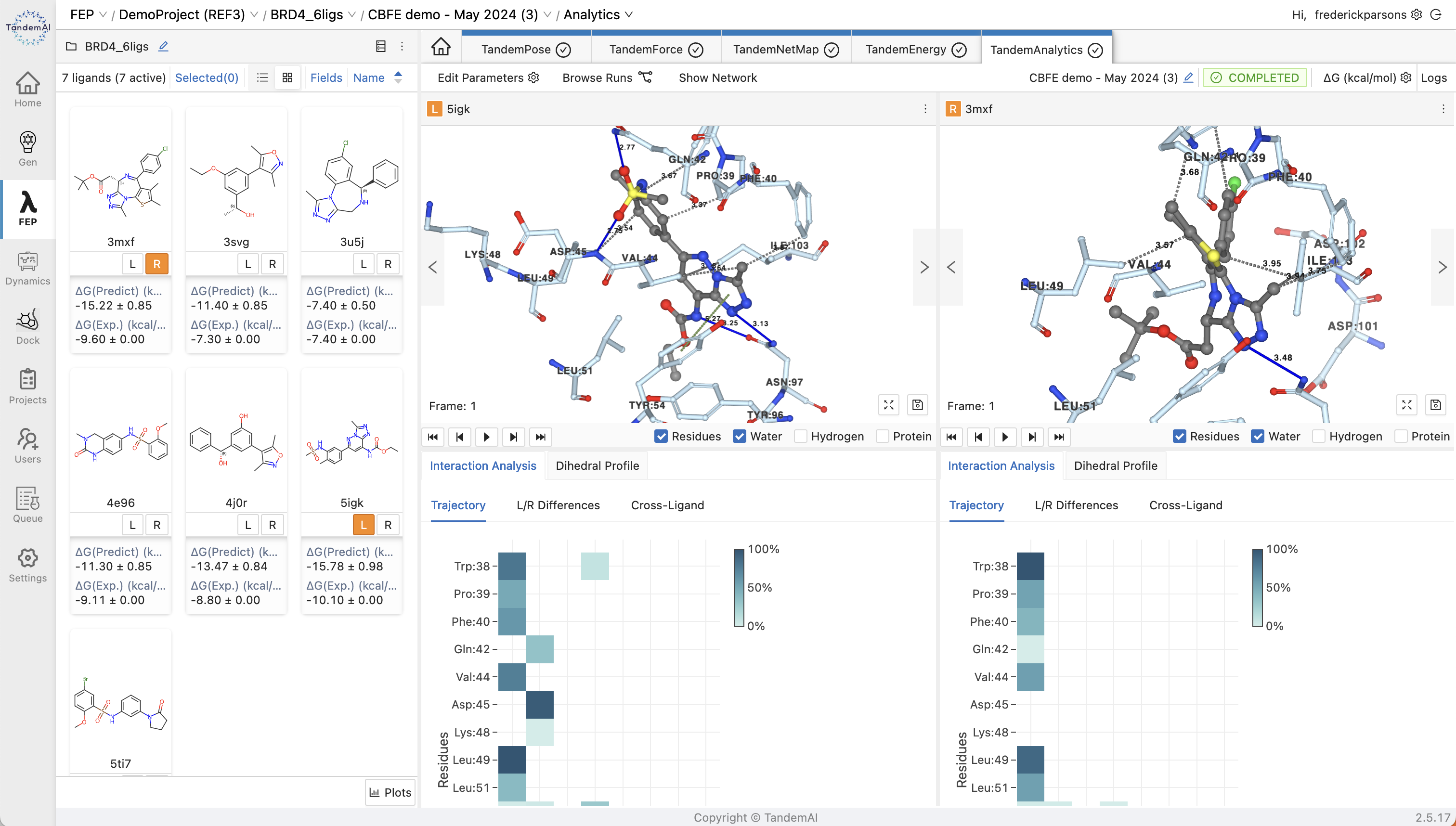
Within TandemFEP, you can use our TandemAnalytics interface to quickly identify the best candidates for your project through interaction analysis visualizations. You can also explore your molecules further with tools like TandemPose to investigate new starting conformations.
Incorporating TandemAI’s core hopping workflow into your drug discovery process can significantly accelerate the identification and optimization of your next therapeutic candidate. By leveraging our advanced CBFE technology and integrated platform tools, you can explore new chemical spaces with unprecedented efficiency and accuracy, opening the door to unlimited possibilities. Streamline your path to success by making TandemAI part of your team today.
Watch our webinar to learn more about the combined power of core-hopping with CBFE
Explore our drug discovery solutions