Combining the precision of small molecules with the specificity of larger biologics, peptides are rapidly emerging as a therapeutic modality. Their wide variety and diversity open new avenues of opportunity for drug discovery teams, a prospect that is particularly promising for challenging targets where small molecules have proved ineffective. Despite their potential, the industry still faces considerable hurdles in successful peptide therapeutic discovery, including the successful identification of high-quality hits, efficient optimization of binding affinity, and improving membrane permeability and other critical drug-like properties.
Predictive computational tools that have aided in these challenges with traditional small molecule drugs have proved elusive for peptide therapeutics due to the greater complexity of larger biological systems. However, recent advances in the field have led to novel methods with the potential to greatly reduce the time and cost of hit finding and optimization, accelerating the discovery of therapeutic peptides.
Peptides cover an extensive chemical space across canonical and non-natural amino acid combinations, and various chemical linkers, making the identification of high-quality starting points tremendously challenging. Since traditional wet lab methods for hit identification rely heavily on empirical screening and limited chemical libraries, this approach can be especially inefficient for peptides and often result in resources being wasted on lower-quality candidates.
The emergence of generative approaches provides the opportunity to explore much larger chemical spaces virtually, without investing in larger libraries and screening campaigns. By leveraging de novo generative design in TandemGen, you can rapidly generate novel peptide binders, custom-fit to your target of interest, to quickly identify the best possible peptide candidates and advance them to the next stage of development.
Optimizing the binding affinity and selectivity of peptides is a key component of early discovery, often requiring a large investment in slow and inefficient iterative methods. With our platform, your team can evaluate hundreds of ideas per week without the time and costs associated with peptide synthesis and in vitro testing, enumerating large libraries of peptide analogs around a single hit in TandemGen and accurately calculating their binding affinity through our custom-developed Free Energy Perturbation (FEP) engine in TandemFEP .
Using our platform, we generated several novel peptide inhibitors that modulate a cancer target of interest in less than 18 months. In this program, we generated hundreds of peptide analogs in TandemGen and seamlessly evaluated their affinity to the protein of interest in TandemFEP within a single, integrated workflow. The program demonstrated consistently accurate affinity predictions across the entire series (Figure 1) and successfully identified a low nanomolar peptide as a pre-clinical candidate.
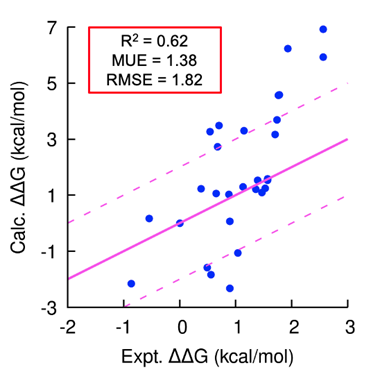
Figure 1: Accurate prospective peptide FEP predictions; peptide designs involved mutations to both canonical and non-canonical amino acids and contained chemical linkers
For intracellular targets, membrane permeability is a critical aspect of peptide therapeutic design. However, due to the varied charge, hydrophilicity, and size of amino acids, achieving optimal permeability for peptides is a defining challenge.
With our advanced physics-based and machine-learning permeability models, you can screen your candidates for membrane permeability as part of your optimization workflow. Combined with the power of other tools like TandemADME and TandemFEP , you can rapidly generate, model, and evaluate peptide structures, chemical linkers, and modifications to identify hits that strike a perfect balance between permeability and potency within hours.
The inherent low permeability and instability of peptides present a distinct challenge for the optimization of critical ADME parameters, creating a pressing need for accurate predictive tools to help drive affinity while maintaining a necessary level of cell permeability and metabolic stability. The use of computational tools to help drive the multiparametric optimization of peptides during a discovery program is critical to progress peptide therapeutics into the next stage of development.
With TandemAI’s fully integrated platform, you can access all the AI and physics-based modeling you need to refine your peptide hits, seamlessly move between tools, and connect wet lab experiments within a single, collaborative, web-based portal. Modulate drug-like properties in TandemADME , perform molecular dynamics in TandemDynamics , and model molecular interactions in TandemDock . By enabling you to optimize multiple parameters at once, TandemAI delivers efficient fine-tuning of your peptide candidates in a fraction of the time.
By significantly shortening the time to candidate discovery, TandemAI’s innovative computational solutions are revolutionizing the field of peptide therapeutics. Our technologies tackle the fundamental challenges of hit discovery, binding optimization, membrane permeability, and structural stability, streamlining the discovery process. TandemAI's AI and physics-based approaches can put you at the forefront of a new wave of peptide drug development, paving the way for advanced therapeutic solutions.
Explore our full range of computational drug discovery tools today.