Regulatory hurdles, patent issues, market pressures, and high candidate failure rates are only a few of the challenges faced by drug discovery companies. For smaller companies and startups, this endeavor can be particularly daunting, with the early stages of the drug discovery process draining time and resources. By leveraging generative AI and free energy perturbation (FEP) calculations, researchers can accelerate their hit identification, reduce their timelines, and increase their return on investment. At TandemAI, we combined these tools and more into one convenient platform, to streamline your path to success.
Most AI drug-discovery tools are designed for expert use and run disconnected from each other in a local environment, creating a time sink where generated data needs to be moved across platforms and programs.TandemViz eliminates this time sink by integrating a comprehensive range of early-stage drug-discovery tools into a single, user-friendly interface, allowing you to move from one stage of development to the next quickly and easily.
With TandemViz, you can generate novel ideas through a range of generative and reaction-based enumeration methods in TandemGen and move seamlessly to assessing their binding affinity to a target in TandemFEP.
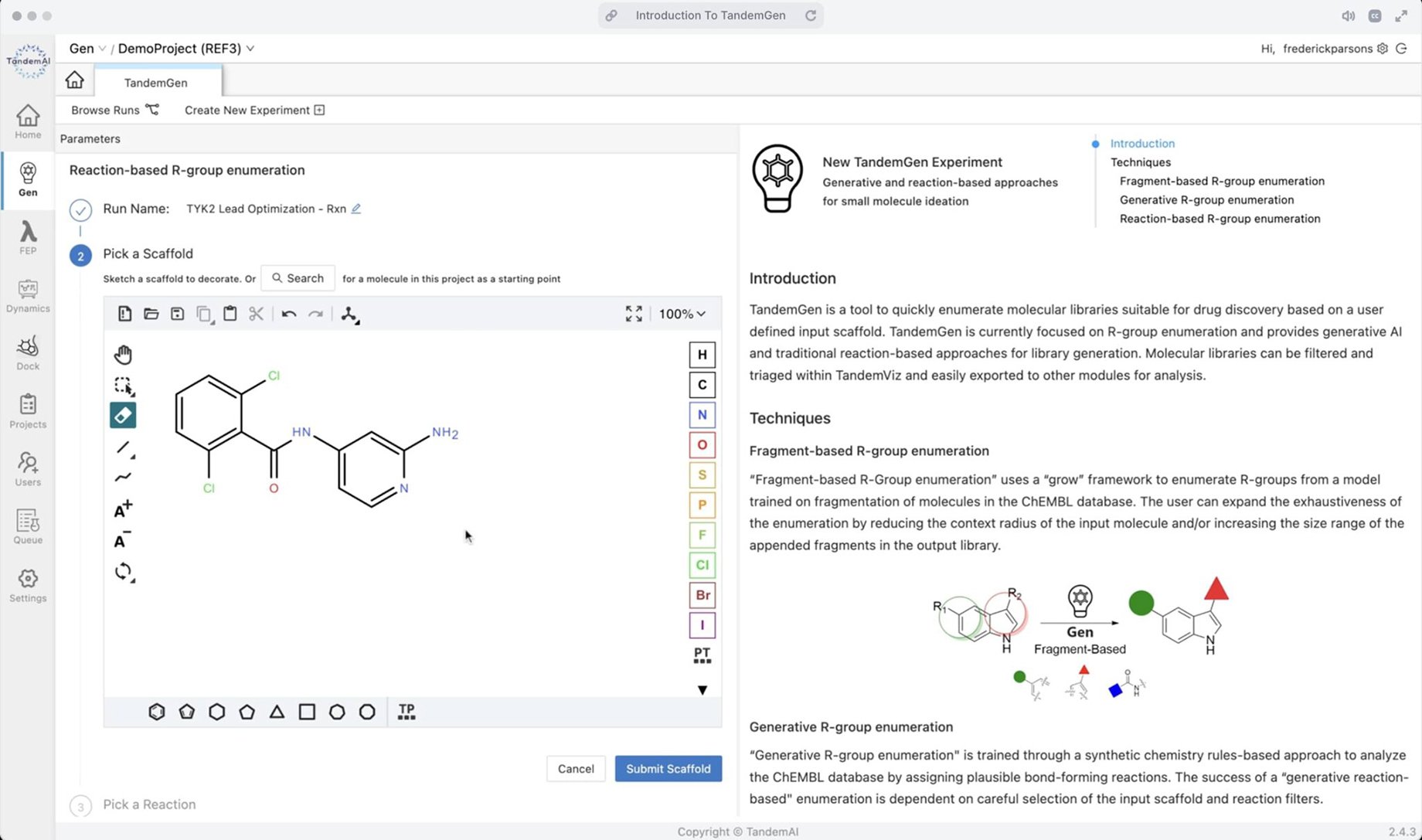
Quickly and easily create your TandemGen run by first selecting the generation technique:
| Generative R-Group enumeration | Generating analogs of a specific molecule from a library of R-group fragments. |
|---|---|
| Fragment-based enumeration | Building small molecules up from molecule fragments. |
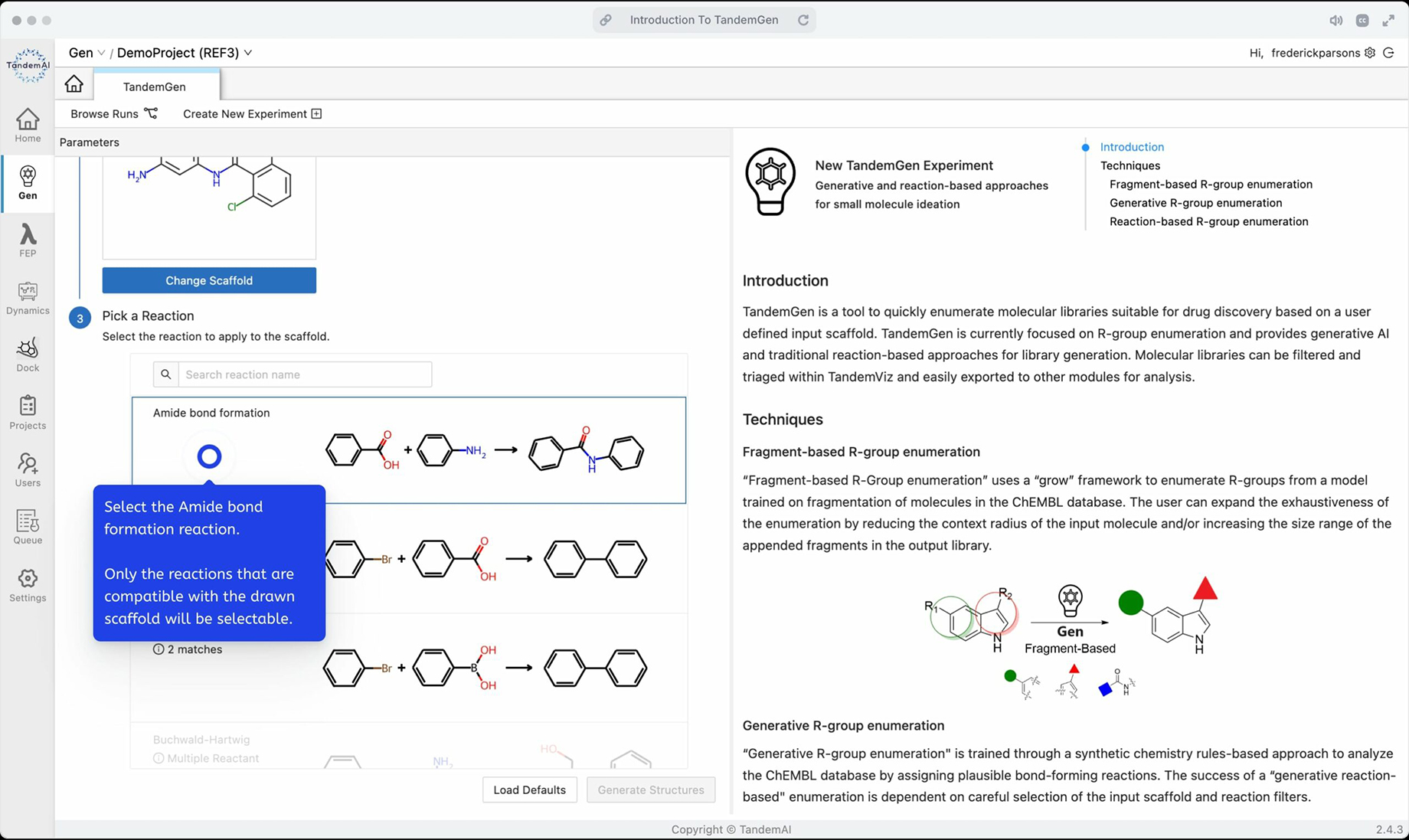
| Reaction-based enumeration | Using reaction templates to generate a library of molecules capable of participating in the modeled reaction. |
| Core-Hopping Enumeration | Identifies alternative cores of roughly similar size that maintain similar R-groups orientations |
You can then pick a scaffold from the project to decorate or draw your own in the sketcher, and choose a reaction to apply to complete your query.
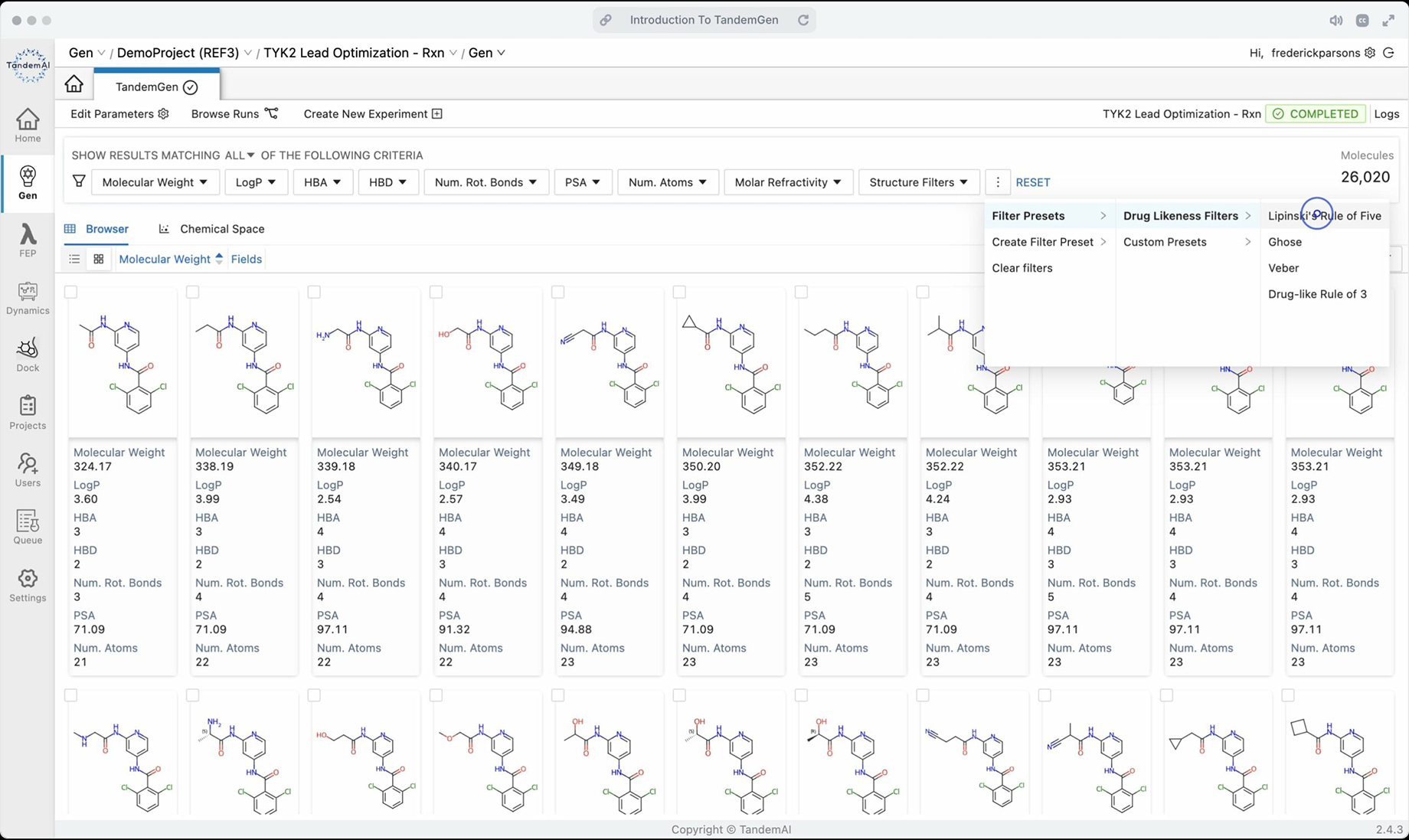
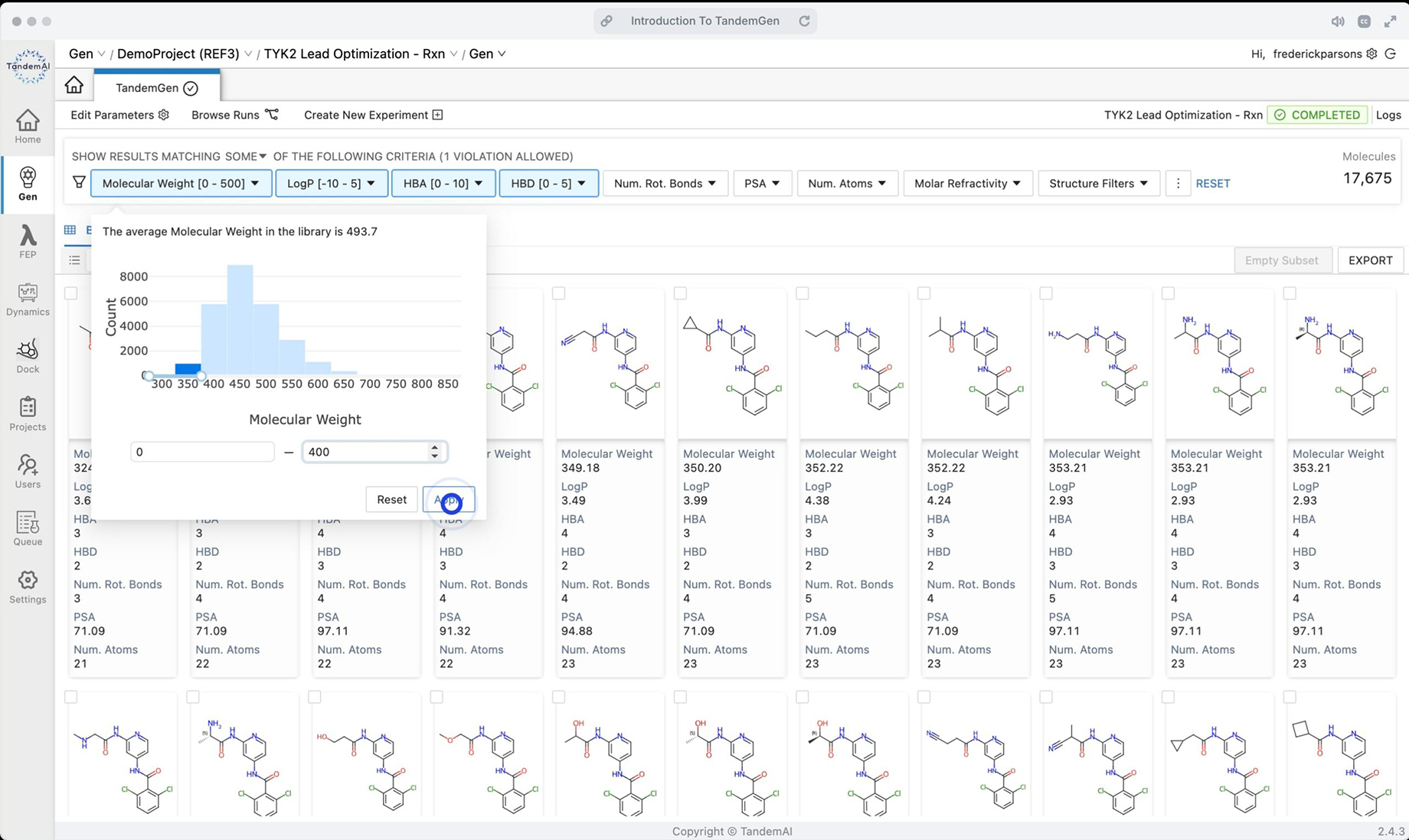
A vast library of synthetically accessible candidates is generated instantly from your query, opening up your research space with limitless possibilities. You can explore your candidates by utilizing TandemFilters, designed by our medicinal chemists, to ensure they are chemically reasonable and fit your specific design goals.
Gain even deeper insights into your data in the chemical space with t-distributed stochastic neighbor embeddings (t-SNE). By leveraging this feature you can visualize high-level similarities between your candidates and create custom plots to further stratify those similarities by different physical and molecular properties.
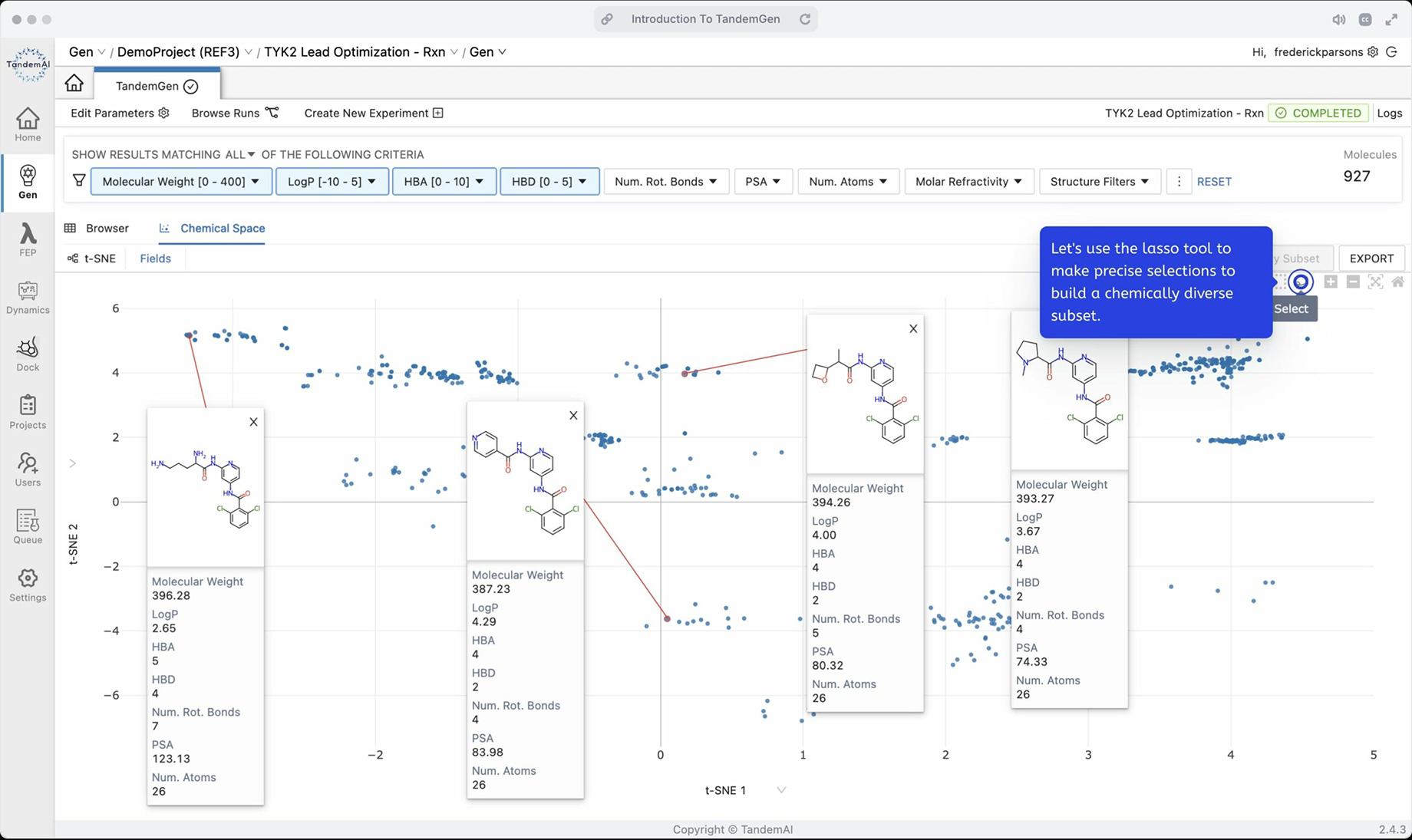
With our integrated platform, you can easily send candidates to other applications in TandemViz, allowing you to move to binding affinity testing in TandemFEP with a click of a button. Compute binding affinities with near-experimental accuracy with the rigorously derived molecule force field parameters used in TandemFEP. This validated FEP workflow also assists you in deriving rationales for different molecule potencies through its extensive analytics interface that combines computational and experimental data.
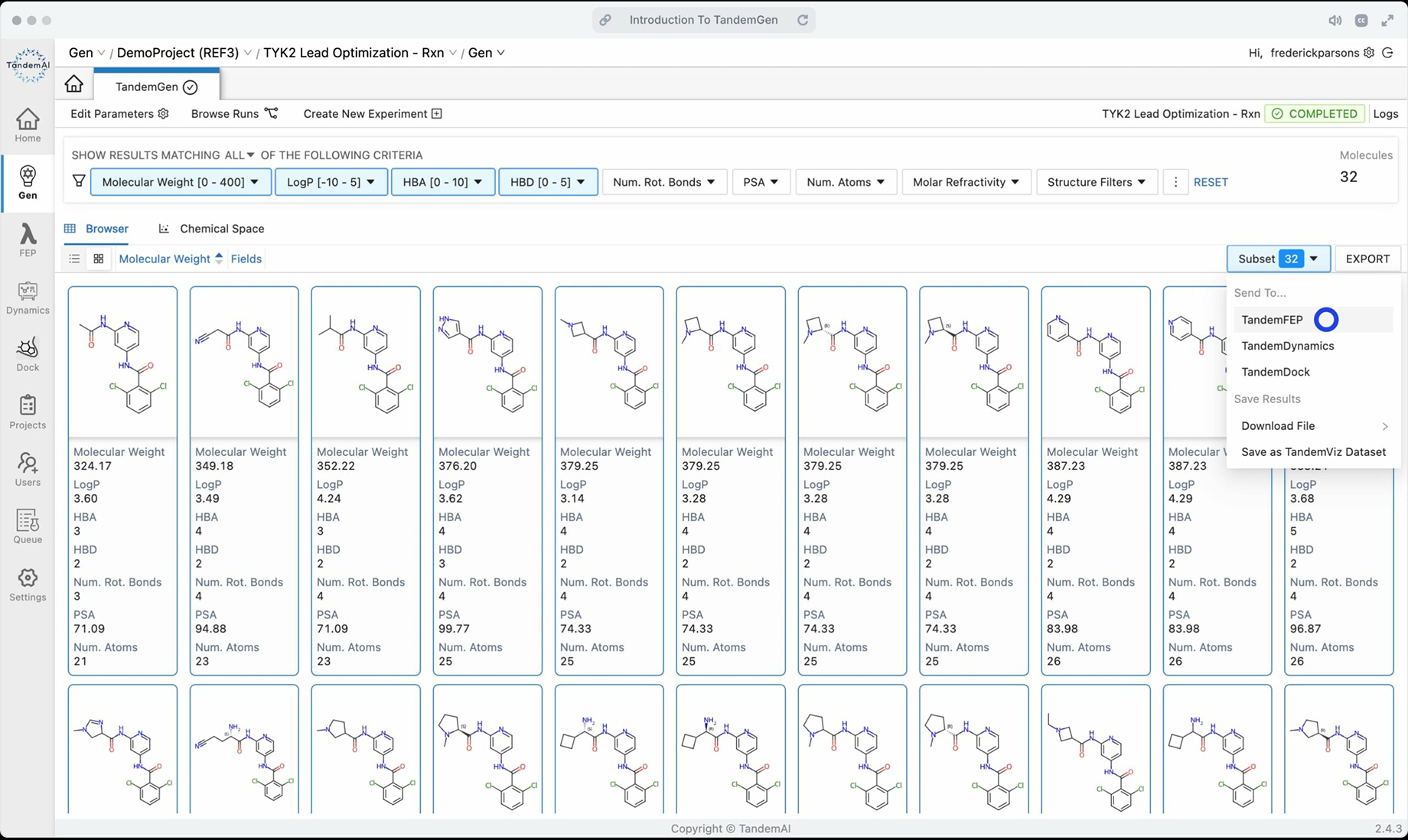
With so many demands on the time and financial resources of drug discovery teams, utilizing tools that reduce these burdens is essential for success. With TandemAI, there’s no need to find and license interoperable drug discovery software, TandemViz brings you all the tools you need in one integrated interface. Leverage our TandemGen to TandemFEP pipeline to focus your research efforts on compounds with the highest probability of potency and expedite your successful discoveries today.