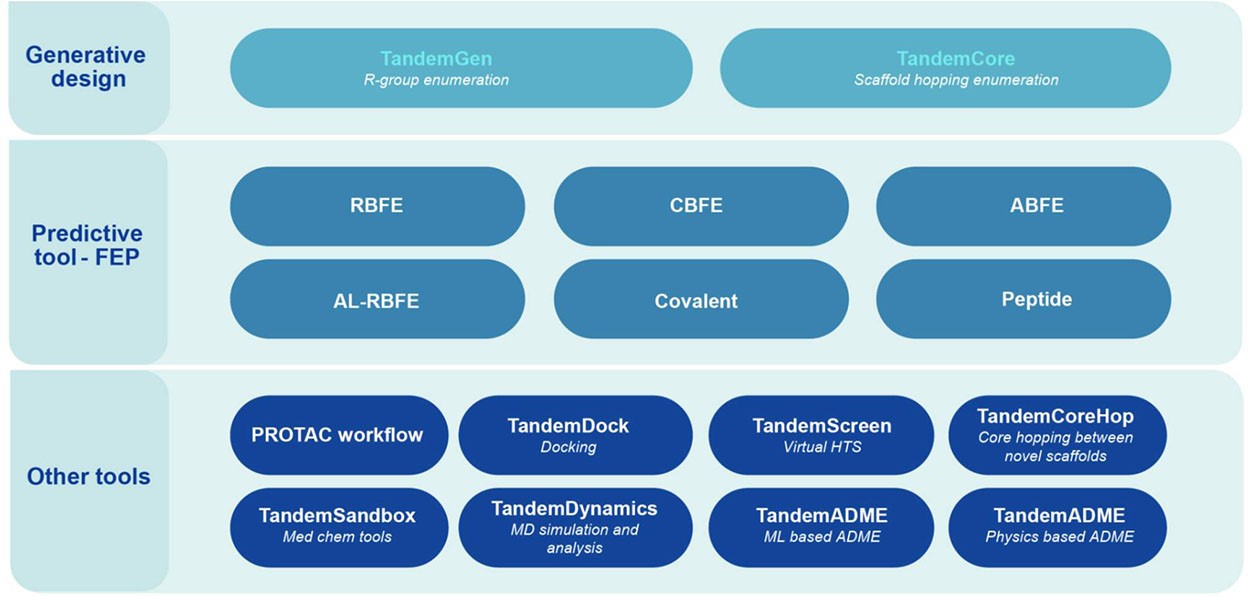
Leveraging industry-leading physics-based and AI algorithms in our drug discovery platform with a full suite of computational tools designed to accelerate your hit-to-lead progression and lead optimization.
Available through an FTE partnership or SaaS license, with optional GPU resources, virtual private cloud (VPC) deployment, and expert application support included, we curate the relationship your team needs - our services, your way.
Accurately compute binding affinities
Compute protein-small molecule binding affinities to near experimental accuracy using our free energy perturbation (FEP) potency assay, TandemFEP, which utilizes small molecule force field parameters rigorously derived from quantum mechanics calculations.
Predict ADMET with precision
Predict absorption, distribution, metabolism, excretion, and toxicity (ADMET) endpoints for all small molecule candidates using TandemADMET, built on machine-learning models with industry-leading accuracy.
Quickly enumerate novel candidates
Utilize the generative AI and library-based small molecule ideation in TandemGen to rapidly enumerate high-quality and novel small molecule candidates. Predict with confidence using our meticulously curated feature, TandemFilters, designed by our medicinal chemists, to ensure our generated molecules are chemically reasonable.
Gain insights from molecular dynamics simulations
Perform atomic-level molecular dynamics simulations of proteins and small molecules with TandemDynamics. Leverage the interactive 3D viewer to visualize your simulations and gain crucial insights into conformational changes and binding interactions.
Visualize molecular interactions
Model and visualize molecular interactions between proteins and small molecules in TandemDock equipped with an intuitive, interactive 3D-viewer.
Analyze structure-activity relationships
Combine computational and experimental data with our powerful analytics interface, TandemAnalytics, to derive dynamic structure-activity relationships for small molecules at the atomic level.
Collaborate on structural modeling
Create and share structural models and small molecule designs in TandemSandbox. Explore structural possibilities if full with a range of applications from protein and ligand preparation to advanced predictive tools like binding site detection and protein structure prediction.
Learn more about our computational drug discovery tools
Get in TouchLorem ipsum dolor sit amet, consectetur adipiscing elit. Curabitur quis risus id enim vehicula interdum et ac sem. Pellentesque habitant morbi tristique senectus et netus et malesuada fames .
Ac turpis egestas. Integer varius arcu in eleifend elementum. Integer accumsan, libero vel tempor feugiat, sem ligula efficitur dui, sit amet convallis dui purus sit amet orci.
Sed varius efficitur ex sed suscipit. Etiam sit amet arcu id quam convallis vestibulum. Nullam sit amet nisi sit amet ipsum eleifend ultricies quis et purus. Maecenas interdum lobortis maximus.
Compute protein-small molecule binding affinities to near experimental accuracy using our free energy perturbation (FEP) potency assay, TandemFEP, which utilizes small molecule force field parameters rigorously derived from quantum mechanics calculations.
Predict absorption, distribution, metabolism, excretion, and toxicity (ADMET) endpoints for all small molecule candidates using TandemADMET, built on machine-learning models with industry-leading accuracy.
Utilize the generative AI and library-based small molecule ideation in TandemGen to rapidly enumerate high-quality and novel small molecule candidates. Predict with confidence using our meticulously curated feature, TandemFilters, designed by our medicinal chemists, to ensure our generated molecules are chemically reasonable.
Perform atomic-level molecular dynamics simulations of proteins and small molecules with TandemDynamics. Leverage the interactive 3D viewer to visualize your simulations and gain crucial insights into conformational changes and binding interactions.
Model and visualize molecular interactions between proteins and small molecules in TandemDock equipped with an intuitive, interactive 3D-viewer.
Combine computational and experimental data with our powerful analytics interface, TandemAnalytics, to derive dynamic structure-activity relationships for small molecules at the atomic level.
Create and share structural models and small molecule designs in TandemSandbox. Explore structural possibilities if full with a range of applications from protein and ligand preparation to advanced predictive tools like binding site detection and protein structure prediction.
Combine computational and experimental data with our powerful analytics interface, TandemAnalytics, to derive dynamic structure-activity relationships for small molecules at the atomic level.
With access to state-of-the-art computational tools through our drug discovery platform, you can generate up to 5x more high-quality ideas per week and use up to 90% less time and resources to reach your goals.
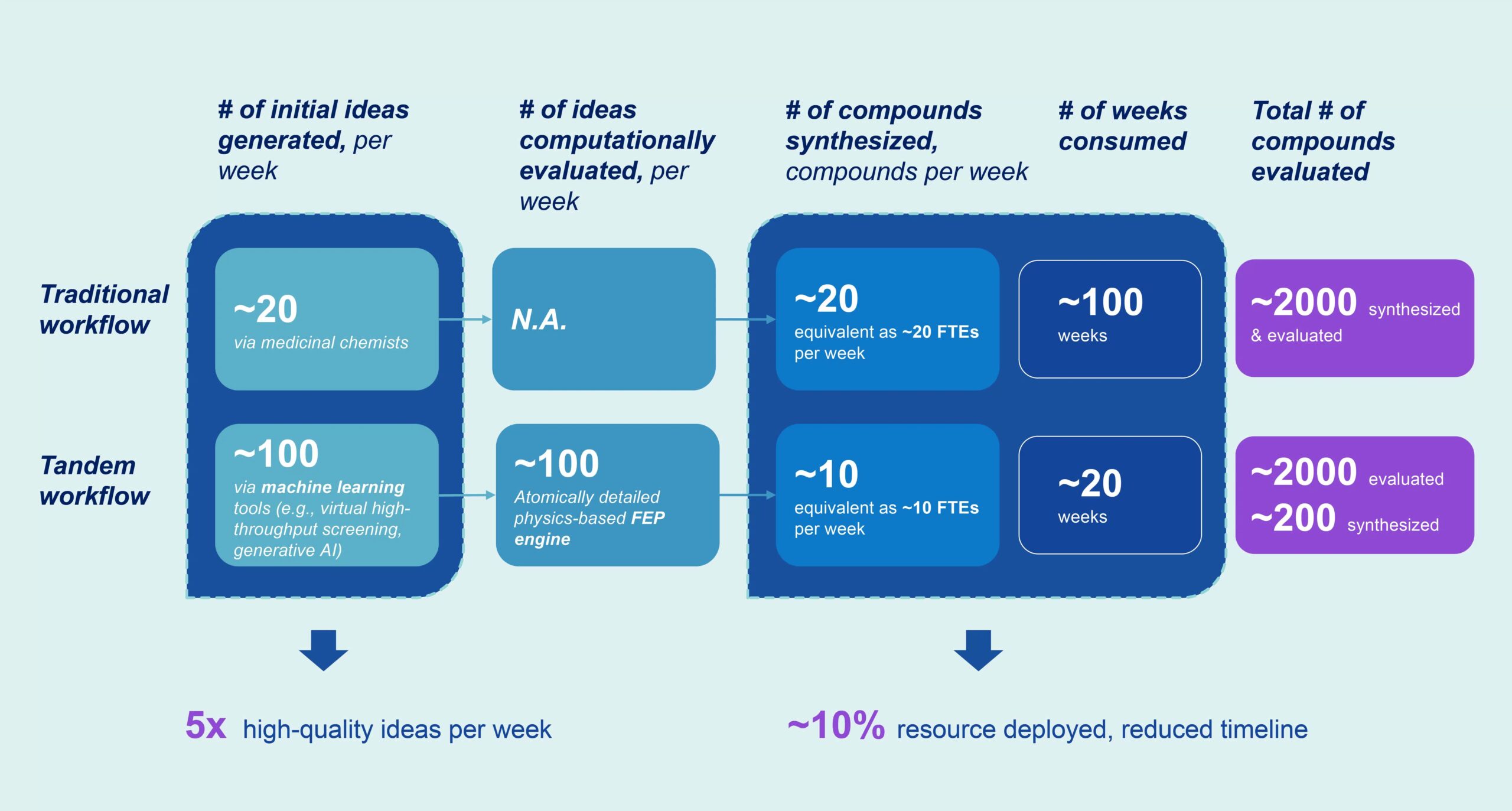
Available by FTE, where you pay for wet lab services and gain complimentary access to our proprietary computational tools or software license (SaaS), with optional GPU services, virtual private cloud (VPC) deployment, and expert application support.
Get in Touch